Highly accurate protein structure prediction with AlphaFold

סקירה זו היא חלק מפינה קבועה בה אנו סוקרים מאמרים חשובים בתחום ה-ML/DL, וכותבים גרסה פשוטה וברורה יותר שלהם בעברית. במידה ותרצו לקרוא את המאמרים הנוספים שסיכמנו, אתם מוזמנים לבדוק את העמוד שמרכז אותם תחת השם deepnightlearners.
לילה טוב חברים, היום אנחנו שוב בפינתנו deepnightlearners עם סקירה של מאמר בתחום הלמידה העמוקה. היום נבחר לסקירה המאמר שנקרא:
Highly accurate protein structure prediction with AlphaFold
פינת הסוקר:
המלצת קריאה מאופיר: קריאה מרתקת, במיוחד למי שמתעניין גם בביואינפורמטיקה. כמות המשאבים שהושקעו במחקר והתוצאות שלו מסחררות. מעבר לחידושים עבור הבעיה הספציפית, מוצגות טכניקות חדשות באופן כללי.
בהירות קריאה: בינונית-גבוהה.
רמת היכרות עם כלים מתמטיים וטכניקות של ML/DL הנדרשים להבנת המאמר: נדרשת היכרות עם למידה עמוקה. בנוסף, מומלצת מאוד היכרות עם מושגים ביואינפורמטיים.
יישומים פרקטיים אפשריים: מדובר בכלי החזק ביותר כיום לניבוי מבנה של חלבונים, והוא צפוי לשמש רבות לקידום מחקר החלבונים בעולם, בין אם למדע בסיסי ובין אם לתחומים כמו הנדסת חלבונים. בנוגע לשימושים מסחריים – DeepMind שחררה את המודל והמשקולות, אך השימוש במשקולות אסור לשימוש מסחרי.
פרטי מאמר:
לינק למאמר: זמין כאן
לינק לקוד: זמין להורדה
פורסם בתאריך: 15/07/2021
הוצג בכתב העת: Nature
תחומי מאמר:
- ניבוי מבנה תלת מימדי של חלבונים.
כלים מתמטיים, טכניקות, מושגים וסימונים:
- רשתות נוירונים גרפיות (GNNs).
- Attention.
- Skip-connections.
מבוא והסבר כללי על תחום המאמר:
חלבונים הם הגורמים האחראים על מירב הפעולות שאנו קוראים להן "חיים". החלבונים נוצרים בתהליך תרגום על ידי ריבוזומים מרצף RNA. רצף ה-RNA עצמו ממועתק מרצף DNA ברוב המקרים, למעט אצל וירוסים מסוימים.
המורכבות האדירה של חלבונים מתבטאת בכך שיש להם 20 סוגים של אבני בניין (20 סוגי חומצות אמינו), וכך שחלבונים הם בעלי אורך של עשרות עד אלפי חומצות אמינו. נכון להיום, מוכרים מעל שני מיליארד חלבונים. פרדוקס לוינת'ל מתאר את המורכבות הזו כך – בהינתן המספר העצום של קונפורמציות – מבנים תלת מימדיים – שחלבון יכול להיות בהן, איך הוא מצליח להתכנס לקונפורמציה אחת (או מספר מצומצם) בזמן קצר? כהערת אגב, בנוסף ל-20 אבני הבניין הבסיסיות הנ"ל קיים מגוון רחב של מולקולות העונות להגדרה של חומצות אמינו. חומצות אמינו נוספות אלו עשויות להיות חלק מחלבון בעקבות תהליך של הרחבת הקוד הגנטי, בין אם מדובר במגוון מודיפיקציות שנוספות על חומצת אמינו בחלבון או נסיונות שימוש בחומצות אמינו חדשות בהנדסת חלבונים.
בגלל החשיבות הגבוהה של חלבונים, חשוב לנו לדעת איך הם פועלים עבור מגוון מטרות. לדוגמה:
- להבין איך מנגנונים ביולוגיים עובדים.
- פיתוח תרופות וחיסונים.
- מגוון שימושים תעשייתיים.
בשביל לדעת איך עובד חלבון מסוים אנו צריכים להבין קודם כל את המבנה שלו. בהמשך לפרדוקס לוינת'ל, קשה מאוד לדעת עבור רצף כלשהו של חומצות אמינו מה המבנה התלת מימדי של החלבון. קיימות שיטות פיזיקליות לקבוע מבנה של חלבון, אבל הן דורשות זמן ומשאבים משמעותיים (לפעמים שנים עבור חלבונים חדשים), יקרות מאוד ולא בהכרח מצליחות. דהיינו, אין אופציה ל-scale בצורה הזאת. לכן, יש צורך לנסות לחזות את המבנה בצורה חישובית – משימה שהתבררה במשך השנים כמורכבת מאוד.
חברת DeepMind מבית גוגל הוכיחה שוב את תרומתה של התעשייה למחקר בסיסי, והביאה פתרון לבעיה שעוקף היום בפער ניכר את שאר הפתרונות שהגיעו מהאקדמיה (גילוי נאות – מעבדת המחקר שלי בלימודי התואר השני עוסקת בין היתר בנושא זה).
תמצית המאמר:
המערכת של AlphaFold2 (שהם קוראים לה פשוט Alphafold ולכן נעשה זאת גם כן) מורכבת מאוד ומכילה כמות נכבדת של ארכיטקטורות חדשות שנבנו במיוחד עבורה. נפרק את המערכת בהדרגה לחלקי הבסיס שלה. Alphafold כוללת 5 מודלים נפרדים הפועלים בצורה של ensemble.
ארכיטקטורה כללית:
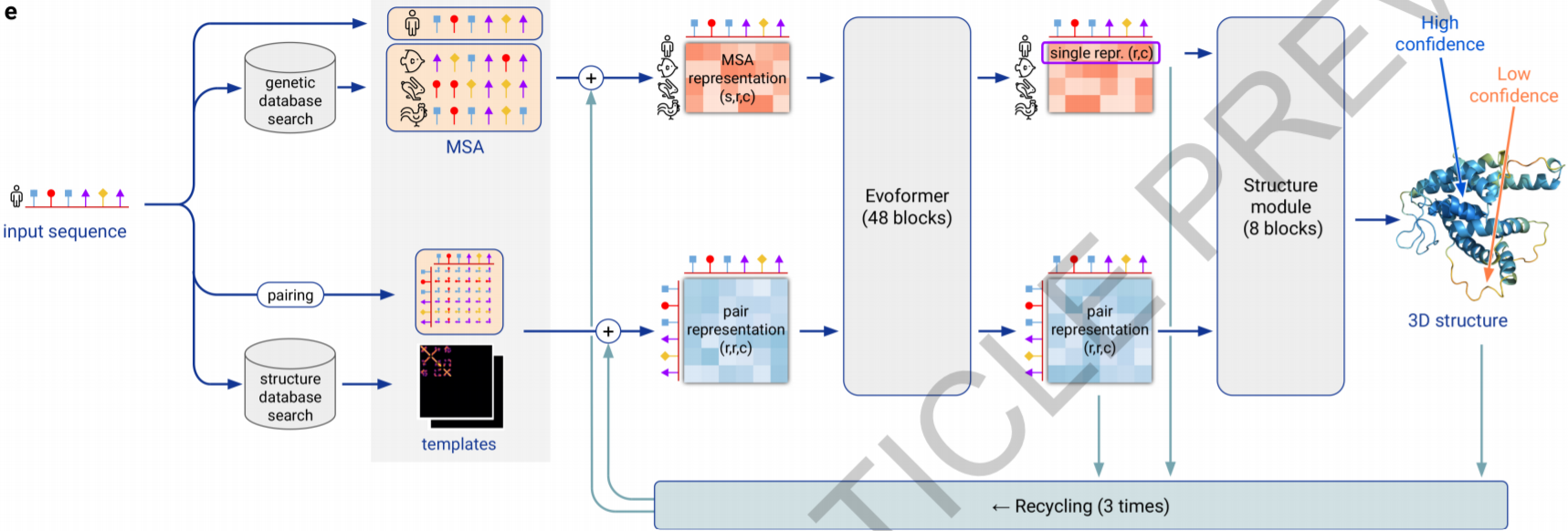
AlphaFold מורכב משני ייצוגים וקטוריים שונים שמתעדכנים במקביל:
- ייצוג MSA – מעמידים רצפים של חלבונים שדומים לחלבון שמעניין אותנו ואת אותו החלבון, בצורה שמייצגת הכי טוב את הדמיון ביניהם על ידי מיקסום של ניקוד. ב-MSA כל שורה מייצגת חלבון שונה, וכל עמודה מייצגת מיקום זהה בתוך החלבונים. העמדה נחשבת עם ניקוד גבוה יותר ככל שיש בה פחות רווחים וחומצות אמינו שונות בעמודה – שמייצגת מיקום משותף לכל החלבונים בהעמדה. האלגוריתם משתמש במאגר מידע של רצפים בשביל ליצור העמדה עבור החלבון.
- ייצוג זוגות – מייצגים מבנה בצורה לטנטית בעזרת מטריצה שמורכבת מהמרחקים בין כל זוג חומצות אמינו בחלבון. בייצוג כזה צריך לוודא כי המרחקים מכבדים את החוקים של מערכת תלת מימדית תקינה (למשל, קיום אי-שוויון המשולש).
שני הייצוגים עוברים יחד תהליך כדלקמן:
- מעבר דרך 48 שכבות של Evoformer.
- לאחר מכן השורה שמייצגת את החלבון שלנו מה-MSA וייצוג המבנה עוברים דרך 8 שכבות מבנה.
- המבנה שמתקבל עובר fine tuning ליצירת המבנה הסופי.
- לבסוף, לאחר העברת הייצוגים דרך שכבות ה-Evoformer והמבנה הם עוברים "מיחזור" (skip connection הפוך, מתווסף לייצוג מהשלב לפני ה-Evoformer) שלוש פעמים.
Evoformer:
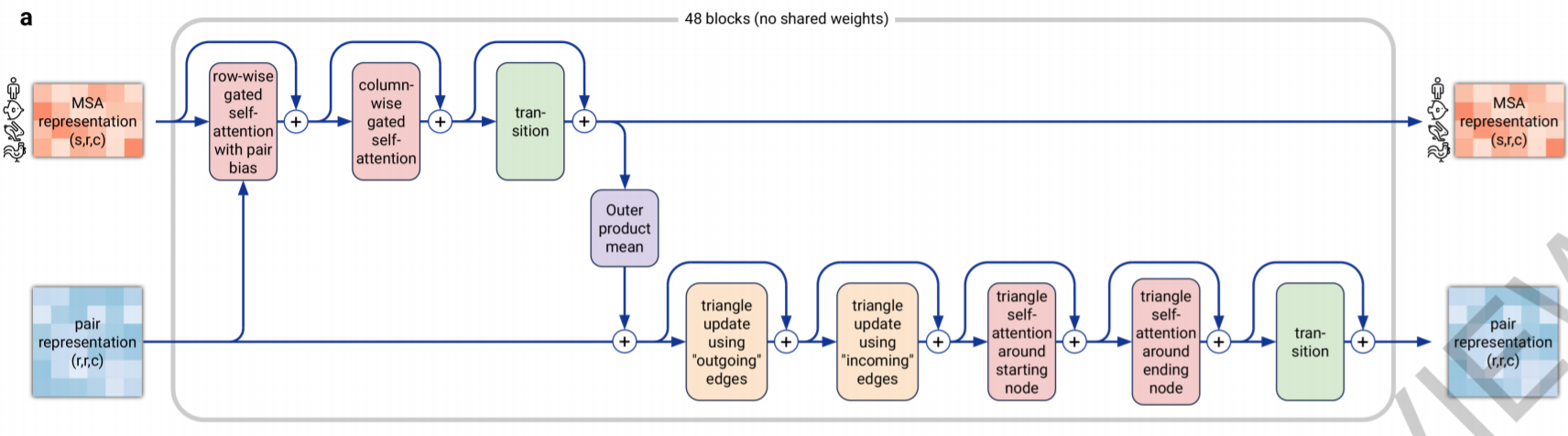
שכבת ה-Evoformer מורכבת ממספר תתי-שכבות אשר ביניהן יש skip-connections.
- ייצוג ה-MSA עובר דרך השכבות הבאות:
- שתי שכבות gated self-attention יחד עם ייצוג הזוגות בתור bias לפי השורות.
- שתי שכבות gated self-attention לפי העמודות.
- לבסוף, הייצוג עובר שכבת transition אשר מעדכנת את הסיבובים וההסטות כדי להפוך את הפעולות לאקוויוריאנטיות במרחב (להסבר על אקוויוריאנטיות במרחב ניתן לקרוא את הסקירה הזו בנושא).
- ייצוג הזוגות מטופל באופן הבא:
- מתווסף אליו ממוצע המכפלה החיצונית של ייצוג ה-MSA אחרי ה-transition.
- לאחר מכן הוא עובר עדכונים לפי שלשות קודקודים (חומצות אמינו) בו.
- לאחר זאת, הוא עובר שכבות self-attention לפי שלשות קודקודים בו.
- לבסוף, הוא עובר שכבת transition בדומה לייצוג ה-MSA.
שכבת מבנה:

שכבת המבנה מזיזה ומסובבת את החומצות האמיניות בחלבון, כאשר העדכון מתבצע לפי שלשות של חומצות אמינו צמודות בחלבון. השורה ב-MSA שמייצגת את החלבון שלנו מתעדכנת בעזרת invariant point attention עם skip connection שפועל עליה, על ייצוג הזוגות ועל שלשות חומצות האמינו.
כעת, תוצאה זו משמשת לעדכון ההסטות והסיבובים של שלשות חומצות האמינו הצמודות. ייצוג הזוגות אינו מתעדכן בשכבה זו.
Fine-tuning:
בסוף התהליך, המבנה המתקבל עובר תהליך fine-tuning פיזיקלי על ידי כלי שנקרא Amber על מנת לטפל בסתירות מבניות כמו התנגשויות מיקומים.
חישוב ה-loss:
Alphafold כוללת מספר סוגי loss ששילובם יוצר את ה-loss הכולל של המערכת.
- מבצעים דיסקרטיזציה על ייצוג הזוגות הסופי ומבצעים עליו cross-entropy loss.
- מבצעים masking רנדומלי על ייצוג ה-MSA ההתחלתי וגורמים לרשת לשחזר את העמדות האלה בעזרת loss בדומה ל-loss של BERT.
- מבצעים loss נוסף על המרחקים הלוקאליים בין פחמני אלפא של השרשרת המרכזית של חומצות האמינו.
- בתהליך ה-fine-tuning הסופי משתמשים ב-loss על הפרת מגבלות מבניות.
דאטא:
מאגר המידע המרכזי ששימש את האלגוריתם הוא PDB שכולל את כלל מבני החלבונים שמבנם נקבע בצורה פיזיקלית.
כמו כן, הדאטה מ-PDB הועשר במידע אודות רצפי חלבונים (עבור ה-MSA) בעזרת מאגר המידע BFD שכולל מעל 2.2 מיליארד רצפי חלבונים מ-66 מיליון משפחות. העשרת מידע נוספת ש-Alphafold משתמש בה היא שימוש בניבויי מבנה שלו שהוא בטוח באיכות הניבוי שלהם בתור מידע נוסף לאימון.
האלגוריתם נבחן בתחרות CASP14 שנחשבת סטנדרט הזהב בתחום ניבוי מבנה החלבונים. פעם בשנתיים הקהילה מאותגרת לנבא מבנים של חלבונים אשר המבנה שלהם צפוי להיקבע בצורה פיזיקלית בחודשים הקרובים, ובכך מדובר ב-blind test set, ללא אפשרות לרמאות.
הישגי המאמר:
Alphafold מהווה כרגע הכלי החזק ביותר לניבוי מבנה תלת מימדי של חלבונים, בפער רב.

היכולות אותן הוא הציג צפויות לקדם את חקר החלבונים באופן נרחב, מבחינת פונקציונליות, חקר מחלות, תרופות, חיסונים ועוד. DeepMind שחררה את המודל ואף את המשקולות לשימוש חופשי במחקר מדעי.
עם זאת, Alphafold מאוד תלוי בכמות המידע שיש לו על חלבונים קרובים (MSA). החוקרים מציינים כי עבור חלבונים עם פחות מכ-30 חלבונים דומים אליהם, איכות החיזוי יורדת משמעותית. כפועל יוצא, יכולת הגנרליזציה שלו מוגבלת והוא לא יוכל להתמודד בצורה טובה עם חלבונים רנדומליים. כמו כן, הוא אינו יודע כיצד להתמודד עם חומצות אמינו שונות מה-20 הסטנדרטיות, ולכן עדיין אי אפשר להשתמש בו במחקרים הכוללים חומצות אמינו נוספות (הרחבת קוד גנטי).
נ.ב:
מתחולל כעת אירוע דרמטי בקהילה המחקרית של ניבוי מבני חלבונים, עם ההבנה שמעבדות מחקר יתקשו להתחרות בכמות המשאבים עם חברות כמו DeepMind. התחום בטלטלה, וייתכן ותבוטל תחרות CASP בהמשך.
#deepnightlearners
הפוסט נכתב על ידי אופיר יזרעאלב.
אופיר עובד ב-Dell EMC בתור data scientist במקביל ללימודי תואר שני בדאטא סיינס וביואינפורמטיקה באוניברסיטת בן גוריון ומתעניין בדאטא סיינס ב-irregular domains כגון רשתות נוירונים גרפיות ורשתות נוירונים בינאריות.


